
一、 运行meerkat
前面已经依序安装了meerkat 的环境和meerkat,运行了预处理一步,在相对应的bam文件目录下生成了大批文件,因此,当要用meerkat处理某个bam文件时,应先将该bam文件移动到专有的一个文件夹,manual中也建议这样用。
预处理生成的文件包括:
黑名单文件.gz
isinfo文件:包括插入大小信息
pdf文件:插入大小的分布图,unmapped reads长度的分布图,softclip reads长度分布图
pre.log文件:日志文件,包括输入的参数,输入输出信息,reads数,unallign reads数等
softclips.fq.gz: softlip reads文件,完整的read
softclips.rdist:softclip 的reads长度分布信息记录
unmapped.fq.gz:unmapped的reads 文件,完整的read
unmapped.rdist: unmapped的reads长度的分布信息
sr1.fq.gz : 从softclip read 或者 unmaped read 切下来的指定bp 的reads
sr2.fq.gz : 与sr1.fq配对的reads
一个文件夹包括1_1fq.gz,1_2fq.gz , 里面有不同长度的reads。每个read group 人工的reads 对
1.1 meerkat.pl
perl ./scripts/meerkat.pl [options]
-b FIle 已经sort过的bam文件
-k int [0/1]是否使用预处理产生的黑名单文件,默认1
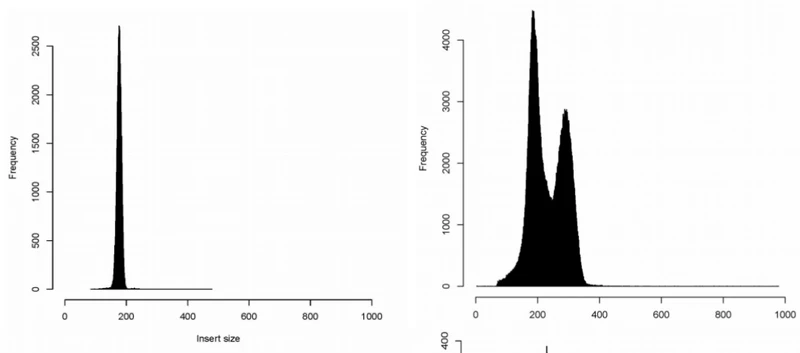
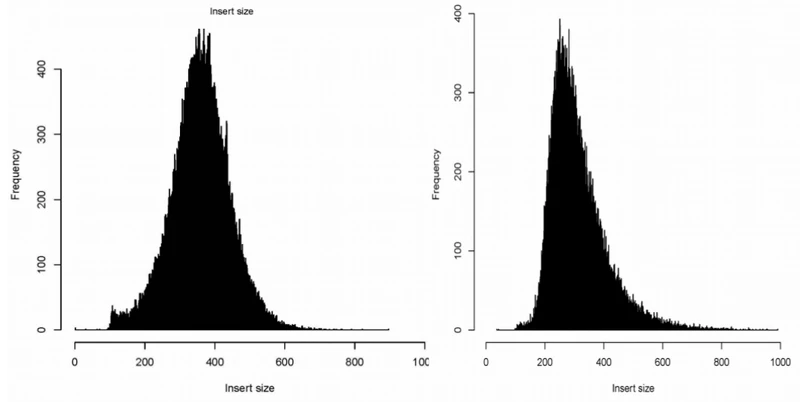
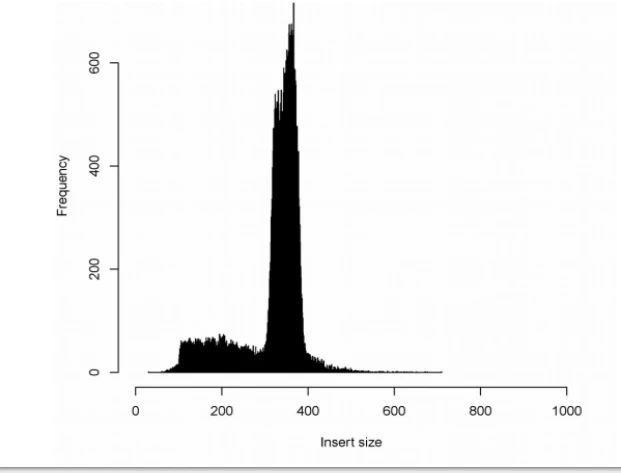
-d FLT call discordant mate pairs 的标准差阈值,默认3.这个选项控制怎样寻找discordant read对,等同于定义最大的concordant fragment(配对的reads定位到的片段),计算方法是:中位插入大小+d x sd,如果插入大小分布图狭窄并对称,就使用默认参数,例如下面一二图。如果分布图偏宽,或者带着长尾,三四图,参数应设为5,对于高深度(>30x),尽管分布图狭窄,但是使用5会轻微好一点。如果峰窄,但是仍然带着一个尾部(图五),好像大部分TCGA数据那样,在meekat.pl这一步使用5比3更好
-c FLT 集簇discordant mate pair标准差阈值,默认和-d相同。控制怎样集簇,构建断点的置信区间。等同于定义覆盖断点的最大片段(中位插入大小+c x sd)。如果-d 选项是5或者小于5,使用用-c相同的参数,如果-d 很大,比如10,那么使用更小的-c,比如5。如果数据有很高的测序深度,或者有广泛的拷贝的变化,那么使用5而不是3来避免不能构建断点的置信区间。
-p FLT call一个事件支持的配对数阈值,默认2
-o INT 需要支持全长reads对的数目,默认是0,设定这个选项会降低小复杂事件的敏感度。如果使用-a 1 -l 1推荐使用-o 1 来避免重复序列导致的过多人工产物
-q INT call一个事件支持的split reads数,默认是1
-z INT 事件数的最大值,默认1,000,000,000
-s INT 从unmapped reads 开始和末端切下来的bp数,必须和与处理的-s 参数设置一样
-m INT [0/1],如果设定为1,使用meerkat 去去除duplicates,如果设为0,使用Picard 的 flag 'd‘ marked ,或者其他工具去除duplicates.bwa mem 的数据必须用picard mark duplicate ,所以要设为0.
-a INT [0/1],处理非单一比对,默认1。如果测序质量不好,或者测序深度不够,将参数设为0,这样全部成对的reads都被作为单一比对使用。这依赖bwa mapping 时生成的XT标签。如果没有XT标签,使用选项Q.
-u INT [0/1],使用bam文件的全部比对,如果BAM文件不是由BWA产生的,或者由bwa产生但是没有XT标签,那么开这个选项,开了这个选项会强制关闭-a选项。默认是0。开了这个选项之后应使用-Q 参数过滤低mapping reads,推荐-Q设置10,对于bwa比对过的bam,也可以继续过滤。
-Q INT 被使用的reads的最小mapping quality,默认是0
-g INT 在主要的bam文件使用的备择mapping数,默认使用全部。备择mapping 数之前通过bwa -N 参数输出到XA标签中。bwa mem 默认 输出备择mapping。
-f INT 在clipped 比对中考虑的备择mapping数,默认使用全部。
-l INT [0/1],是否考虑clipped 比对,默认1.和预处理一样,对于bwa mem 出来的基因组,不需要重新mapping.
-t INT 在bwa比对中用到的核数,默认1
-R STR 包括黑名单read group的文件,每一行一个read group ID
-F STR fasta文件夹路径
-S STR samtools路径,如果samtools不在环境变量中的话
-W STR bwa路径,如果bwa不在环境变量中的话
-B STR blastall 和 formatdb 的路径,如果不在环境变量的话
-P STR 指定运行顺序,dc|cl|mpd|alg|srd|rf|all,默认all,每一步运行都需要上一步的结果
dc: extract discordant read pairs,提取discordant read对
cl: construct clusters of discordant read pairs,将discordant read对建簇
mpd: call events based on read pairs,基于read 对call 事件
alg: align split reads to candidate break point regions,比对split read 到候选的断点区域。如果你有多核心的话,可以将alg步骤切为两步,alg1和alg2,alg1运行多线程,alg2运行单线程。
srd: confirm events based on split reads and filter results,基于split reads和过滤的结果对事件进行证明
rf: refine break points by local alignments,通过区域比对定义断点
all: run all above steps ,运行全部步骤
-h 帮助
manual 中的举例:
50bp reads,<10x 的TCGA 基因组 使用-s 18 -d 5 -a 0 -l 0 -q 1,猜测:reads 长度较小,所以取1/3 read 长度,-s 取18, TCGA 基因组,插入分布狭窄带尾,所以-d 设为5, 测序深度较低,reads 长度较短,所以尽量保留数据,-a 设为0, -l 设为0,-q 设的较低,1。
75-101bp reads, 30-40x TCGA 基因组 使用-s 20 -d 5 -p 3 -o 1 -a 0 -u 1 -Q 10,猜测:reads长度较长,取1/5read长度,-s 取20,TCGA 基因组,插入分布狭窄带尾,所以-d 设为5,长度较长,深度较深,因此降低敏感度,增加特异度,所以-p 设为3,-o 设为1,由于没有XT tag和XA tag ,因此-a 1 选项无法运行,因此设为-u 1 -a 0 -Q 10 。如果这是bwa mem的数据的话,参数应设为-s 20 -d 5 -p 3 -o 1 -m 0 -l 0,bwa mem 数据不需要重新比对softclips -l 0,必须用picard去除duplicate,-m设为0,估计这个是早版本的bwa,因此比对时可以生成XT标签,-a 默认为1。
101bp reads, 60-80x TCGA 基因组 使用-s 20 -d 5 -p 5 -o 3,75-101bp 使用-s 20,TCGA基因组使用-d 5,测序深度深,-o 设置更高3。
如果肿瘤基因组60x,相对应的正常基因组30x,那么使用60x的参数,常常用配对的正常组织中的black.list.gz 重命名并放到肿瘤bam文件处理的文件夹,替换cancer的blacklist.gz文件。
1.2 mechanism.pl
perl ./scripts/mechanism.pl [options]
-b FILE sort过的bam文件
-o INT [0/1]在TE包括rmsk类型 \"Other\",默认1
-t INT TE的最大值,默认100000
-z INT 被处理的SV的数量限制,默认1000000000
-R STR repeat注释路径,能够从UCSC下载
-h help
二、参照
manual中给出的数据,HapMap个体NA18507(42x深度,100bp读长,500bp插入大小),用10核bwa比对花费1.5天和10GB的内存。30x深度的肿瘤数据,要多于两天并且超过30G的内存,如果测序质量不太好,比如很多的嵌合比对和许多非单一mapped的reads,就会花费更多的时间和内存。、
三、结果
运行完预处理和meerkat.pl后,能够得到两个文件.intra.refined.typ.sorted和inter.refined.typ.sorted,运行完mechanism.pl后,会得到.variants文件,否则,就该回去看一下设置是否出现问题。
运行的肿瘤基因组文件的时候,一定要把正常组织的blacklist文件替换肿瘤基因组的blacklist文件,blacklist文件可以在预处理中生成。如果在预处理中出现错误信息“differing read lengths“,没有关系,仅仅是告诉你在一些read group中长度不一致。
四、包含的其他程序
4.1 转换variant 文件到vcf格式
perl ./scripts/meerkat2vcf.pl -i variantfile -o vcffile -H headerfile -F /db/hg18/hg18_fasta/