
什么是哈温平衡?
怎么做哈温平衡检验?
卡方适合性检验! ,一个群体是否符合这种状况,即达到了遗传平衡,也就是一对等位基因的3种基因型的比例分布符合公式:p2+2pq+q2=1,p+q=1,(p+q)2=1.基因型MM的频率为p2,NN的频率为q2,MN的频率为2pq。MN:MN:NN=P2:2pq:q2。MN这对基因在群体中达此状态,就是达到了遗传平衡。如果没有达到这个状态,就是一个遗传不平衡的群体。但随着群体中的随机交配,将会保持这个基因频率和基因型分布比例,而较易达到遗传平衡状态。应用Hardy-Weinberg遗传平衡吻合度检验方法,把计算得到的基因频率代入,计算基因型平衡频率,再乘以总人数,求得预期值(e)。把观察数(O)与预期值(e)作比较,进行χ2检验。病例组和对照组的基因型分布的观察值和预期值差异无显著性(P>0.05),符合遗传平衡定律.
哈温平衡过滤和MAF过滤的区别?
之前,我对这两个概念有点混淆,后来明白过来了。这两个概念一个是对基因频率进行的筛选,一个是对基因型频率进行的筛选。对于一个位点“AA AT TT”,其中A的频率为基因频率,AA为基因型频率。MAF直接是对基因频率进行筛选,而哈温平衡检验,则是根据基因型推断出理想的(AA,AT,TT)的分布,然后和实际观察的进行适合性检验,然后得到P值,根据P值进行筛选。即P值越小,说明该位点越不符合哈温平衡。
两个目的:
- 计算所有位点的哈温检测结果
- 删除SNP中不符合哈温平衡的位点
1. 计算所有位点的HWE的P值
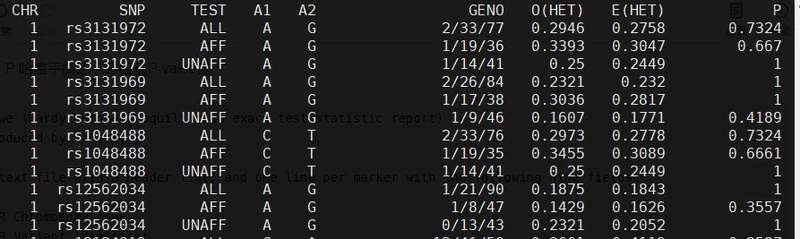
plink --bfile HapMap_3_r3_8 --hardy plink.hwe的数据格式:
- CHR 染色体
- SNP SNP的ID
- TEST 类型
- A1 minor 位点
- A2 major 位点
- GENO 基因型分布:A1A1, A1A2, A2A2
- O(HET) 观测杂合度频率
- E(HET) 期望杂合度频率
- P 哈温平衡的卡方检验P-value值
结果预览:
2. 提取哈温p值小于0.0001的位点
这里我们使用awk:
awk '{if($9 < 0.0001) print $0}' plink.hwe >plinkzoomhwe.hwe 共有123个位点,其中UNAFF为45个位点。
3. 设定过滤标准1e-4
plink --bfile HapMap_3_r3_8 --hwe 1e-4 --make-bed --out HapMap_3_r3_9 日志:
Options in effect: --bfile HapMap_3_r3_8 --hwe 1e-4 --make-bed --out HapMap_3_r3_9 MB RAM detected; reserving MB for main workspace. variants loaded from .bim file. 163 people (79 males, 84 females) loaded from .fam. 112 phenotype values loaded from .fam. Using 1 thread (no multithreaded calculations invoked). Before main variant filters, 112 founders and 51 nonfounders present. Calculating allele frequencies... done. Total genotyping rate is 0.. --hwe: 45 variants removed due to Hardy-Weinberg exact test. variants and 163 people pass filters and QC. Among remaining phenotypes, 56 are cases and 56 are controls. (51 phenotypes are missing.) --make-bed to HapMap_3_r3_9.bed + HapMap_3_r3_9.bim + HapMap_3_r3_9.fam ... done. 可以看到,共有45个SNP根据哈温的P值过滤掉了,和上面手动计算的一样。
4. 可视化
R代码:
hwe<-read.table (file="plink.hwe", header=TRUE) pdf("histhwe.pdf") hist(hwe[,9],main="Histogram HWE") dev.off() hwe_zoom<-read.table (file="plinkzoomhwe.hwe", header=TRUE) pdf("histhwe_below_theshold.pdf") hist(hwe_zoom[,9],main="Histogram HWE: strongly deviating SNPs only") dev.off() 哈温的P值直方图:

过滤掉SNP位点的P值:

过滤后的结果文件
HapMap_3_r3_9.bed HapMap_3_r3_9.bim HapMap_3_r3_9.fam HapMap_3_r3_9.log